Batch jobs#
HPC systems usually are clusters of many compute nodes, which are joined by an interconnect (like InfiniBand (IB) or Omni-PATH (OPA)), and under control of a resource management software (for example SLURM). From the users’ point of view, the HPC system is a unity, a single large machine rather than a network of individual computers. Most of the time, HPC systems are used in batch mode: users would submit so-called “jobs” to a “batch queue”. A subset of the available resources of the HPC machine is allocated to each of the users’ batch jobs, and they run without any need for user intervention as soon as the resources become available.
The job placement, usage monitoring and job accounting are done via a special software, the HPC scheduler. This is an often-under-appreciated automation that makes usage efficient and saves a lot of work on part of the user. However, using HPC is hard in a sense that users have to make an effort in order to figure out what are the available resources on an HPC cluster, and what is the efficient way of requesting the resources for their jobs. Asking for too many resources might be wasteful both in preventing others from using them and in making for a longer queuing time.
The resources (“tractable resources” in SLURM speak) are CPU time, memory, and GPU time. Generic resources can be software licenses, … etc. Requesting resources is done via command line options to job submission commands sbatch and salloc, or via special comment lines or SLURM directives (starting with #SBATCH) in job scripts. There are also options to control job placement such as partitions.
There are default values for the resources which are taken when you do not specify the resource limit. Note that the default values are, as a rule, quite small. On Grex, the default values are set as follow: 3 hours of wall time, 256mb of memory per CPU. In most of the cases, it is better to have an explicit request of an appropriate resource limit rather than using the default.
We ask our users to be fair and considerate and do not allow for deliberate waste of resources (such as running serial jobs on more than one CPU core, or running CPU-only calculations on GPU nodes).
There are certain scheduling policies in place to prevent the cluster from being swamped by a single user. In particular, the MAXPS / GrpRunMins limit disfavors asking for many CPU cores for long wall time, a MaxCPU limits restricts number of CPU cores used, and there are limits on number of user’s jobs in the system and number of array job elements, as described below.
Scheduling policies#
The following policies are implemented on Grex:
- The default wall time is 3 hours (equivalent to: --time=3:00:00 or --time=0-3:00:00).
- The default amount of memory per processor (--mem-per-cpu=) is 2500M. Memory limits are enforced, so an accurate estimate of memory resource (either in the form of --mem= or --mem-per-cpu=) should be provided.
- The maximum wall time is 21 days on genoa, skylake partitions, on largemem and genlm partition.
- The maximum wall time is 7 days on the gpu partition.
- The maximum wall time is 7 days on the preempted partitions: stamps-b, livi-b and agro-b.
- The maximum number of processor-minutes for all currently running jobs of a group without a RAC is 4 M.
- The maximum number of jobs that a user may have queued to run is 4000. The maximum size of an array job is 2000.
- Users without a RAC award are allowed to simultaneously use up to 400 CPU cores per accounting group.
- There are limits on the number of GPUs that can be used on contributed hardware (1 GPU per job).
Note that you can see some information about the partitions by running the custom script partition-list from your terminal:
partition-listTypical batch job cases#
Any batch job is submitted with sbatch command. Batch jobs are usually shell (BASH, etc.) scripts wrapping around the invocation of a code. The comments on top of the script that start with #SBATCH are interpreted by the SLURM scheduler as options for resource requests:
| Directive | Example | Description |
|---|---|---|
| --ntasks= | --ntasks=4 | Number of tasks (MPI processes) per job. |
| --nodes= | --nodes=2 | Number of nodes (servers) per job. |
| --ntasks-per-node= | --ntasks-per-node=4 | Number of tasks (MPI processes) per node. |
| --cpus-per-task= | --cpus-per-task=8 | Number of threads per task (should not exceed the number of physical cores. |
| --mem-per-cpu= | --mem-per-cpu=1500M | Memory per task (or thread). |
| --mem= | --mem=16000M | Memory per node. |
| --gpus= | --gpus=1 | Number of GPUs per job. |
| --time- | --time=0-8:00:00 | wall time in format DD-HH:MM:SS |
| --qos= | * | QOS by name (Not to be used on Grex!). |
| --partition= | --partition=skylake | Partition name: skylake, … etc (very much used on Grex!). |
Assuming the name of myfile.slurm (the name or the extension does not matter, it can be called afile.job, otherjob.sh, … etc.), a job is submitted with the command:
sbatch myfile.slurmor
sbatch [+some options] myfile.slurmSome options like --partition=skylake could be invoked at submission time.
Refer to the official SLURM documentation and/or man sbatch for the available options. Below we provide examples for typical cases of SLURM jobs.
Serial jobs#
The simplest kind of job is a serial job when one compute process runs in a sequential fashion. Naturally, such job can utilize only a single CPU core: even large parallel supercomputers as a rule do not parallelize binary codes automatically. So, the CPU request for a serial job is always 1, which is the default; the other resources can be wall time and memory. SLURM has two ways of specifying the memory: memory per core (--mem-per-cpu=) and total memory per node (--mem=). It is more logical to use per-core memory always; except in case of the whole-node jobs when special value --mem=0 gives all the available memory for the allocated node. An example script (for 1 CPU, wall time of 30 minutes and a memory of 2500M) is provided below.
#!/bin/bash
#SBATCH --time=0-0:30:00
#SBATCH --mem=2500M
#SBATCH --job-name="Serial-Job-Test"
# Script for running serial program: your_program
echo "Current working directory is `pwd`"
# Load modules if needed:
echo "Starting run at: `date`"
./your_program <+options or arguments if any>
echo "Job finished with exit code $? at: `date`"
An important special case of serial jobs is high-throughput computing: jobs are serial because they are too short to parallelize them, however there are very many such jobs per research project. The case of embarrassingly parallel computations like some of the Monte Carlo simulations are often High Throughput Computing (HTC).
- Serial jobs that have regularly named inputs and run more than a few minutes each best be specified as a Job array (see below).
- Serial jobs that are great in numbers, and run less than a few minutes each, better be joined into a task farm running within a single larger job using tools like GLOST, GNU Parallel or a workflow engine like QDO.
An example of GLOST job is under the MPI jobs section (see below).
SMP / threaded / single node jobs#
The next kind of job is multi-threaded, shared memory or single-node parallel jobs. Often these jobs are for Symmetric Multiprocessing (SMP) codes that can use more than one CPU on a given node to speed up the calculations. However, SMP/multithreaded jobs rely on some form of inter-process communication (shared memory, … etc.) that limits them to the CPU cores within just a single server. They cannot scale across multiple compute nodes. Examples are OpenMP, pthreads, Java codes, etc. Gaussian and PSI4 are SMP codes; threaded BLAS/LAPACK routines from MKL (inside NumPY) can utilize multiple threads, … etc. Note that this kind of programs do not scale very well when increasing the number of threads. We recommend to our users to run a benchmark to see how their programs scale with the number of threads to define the combination or a set of threads for better performance.
Thus, from the point of view of the SMP/threaded jobs resources request, the following considerations are important:
- asking always only a single compute node and one task (--nodes=1 --ntasks=1) job.
- asking for several CPU cores on it per job, up to the maximum number of CPU cores per node (--cpus-per-task=N) where N should not exceed the total physical cores available on the node. Depending on the partition, you may choose N up to 52 on the skylake partition, up to 40 on the largemem partition, … etc.
- making sure that the total memory asked for does not exceed the memory available on the node (refer to the section about node characteristics, hardware for more information).
- making sure that the code would use exactly the number of CPU cores allocated to the job, to prevent waste or congestion of the resources.
In SLURM, it makes a difference whether you ask for parallel tasks (--ntasks) or threads (--cpus-per-task) ; the threads should not be isolated from each other (because they might need to use shared memory!) but the tasks are isolated to each own “cgroup”.
An environment variable ${SLURM_CPUS_PER_TASK} is set in the job, so you can set an appropriate parameter of your code to the same value.
For OpenMP, it would be done like:
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}For MKL it is MKL_NUM_THREADS, for Julia --JULIA_NUM_THREADS, for Java -Xfixme parameter.
#!/bin/bash
#SBATCH --time=0-8:00:00
#SBATCH --nodes=1
#SBATCH --ntask-per-node=1
#SBATCH --cpus-per-task=52
#SBATCH --mem=0
#SBATCH --partition=skylake
#SBATCH --job-name="OMP-Job-Test"
# An example of an OpenMP threaded job that
# takes a whole "skylake" node for 8 hours.
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
echo "Starting run at: `date`"
./your-openmp.x input.dat > output.log
echo "Job finished with exit code $? at: `date`"
Note that the above example requests the whole node’s memory with --mem=0 because the node is allocated to the job fully due to all the CPUs anyways. It is easier to use the --mem syntax for SMP jobs because typically the memory is shared between threads (i.e., the amount of memory used does not change with the number of SMP threads). Note, however, that the memory request should be reasonably “efficient” if possible.
It is also possible to use a fraction of the node for running OpenMP jobs. Here is an example asking for 1 task with 4 threads on compute partition:
#!/bin/bash
#SBATCH --time=0-8:00:00
#SBATCH --nodes=1
#SBATCH --ntask-per-node=1
#SBATCH --cpus-per-task=4
#SBATCH --mem=8000M
#SBATCH --partition=skylake
#SBATCH --job-name="OMP-Job-Test"
# An example of an OpenMP threaded job that
# takes 4 threads for 8 hours.
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
echo "Starting run at: `date`"
./your-openmp.x input.dat > output.log
echo "Job finished with exit code $? at: `date`"
GPU jobs#
The GPU jobs would usually be similar to SMP/threaded jobs, with the following differences:
- The GPU jobs should run on the nodes that have GPU hardware, which means you’d want always to specify one of the following options:
| Directive | Description |
|---|---|
| --partition=gpu | to use the gpu partition. |
| --partition=stamps-b | to use the stamps-b partition. |
| --partition=livi-b | to use the livi-b partition. |
| --partition=agro-b | to use the agro-b partition. |
- SLURM on Grex uses the so-called “GTRES” plugin for scheduling GPU jobs, which means that the request syntax in the form --gpus=N or --gpus-per-node=N or --gpus-per-task=N is used.
How many GPUs to ask for?#
Grex, at the moment, does not have GPU-direct MPI enabled, which means that most of the jobs would be single-node. The GPU nodes in either gpu (two nodes, 32GB V100s) or stamps-b (three nodes, 16GB V100s) partition have 4 V100 GPUs, 32 Intel 52xx CPUs and 192GB of CPU memory. There is also the livi-b partition with a large single 16x v100 GPU server. So, asking 1 to 4 GPUs, one node, and 6-8 CPUs per GPU with an appropriate amount of RAM (4-8 Gb) per job would be a good starting point.
Note that V100 is a fairly large GPU for most of the jobs, and for good utilization of the GPU resources available on Grex, it is a good idea to start with a single GPU, and then try if the code actually is able to saturate it with load. Many codes cannot scale to utilize more than one GPU, and few codes can utilize more than two of them.
#!/bin/bash
#SBATCH --gpus=1
#SBATCH --partition=stamps-b
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=6
#SBATCH --mem-per-cpu=6000M
#SBATCH --time=0-12:00:00
#SBATCH --job-name=genomics-test
# Adjust the resource requests above to your needs.
# Example of loading modules, CUDA:
module load cuda/12.4.1
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
echo "Starting run at: `date`"
nvidia-smi
guppy_basecaller -x auto --gpu_runners_per_device 6 -i Fast5 -s GuppyFast5 -c dna_r9.4.1_450bps_hac.cfg
echo "Job finished with exit code $? at: `date`"
The above script (if called say, gpu.job) can be submitted with the usual command:
sbatch gpu.jobDistributed, massively parallel jobs#
Parallel jobs that can spawn multiple servers are the most scalable ones, because they are not limited by the number of CPUs or memory per node. Running many parallel tasks across more than one node requires some inter-node communication (which as a rule is slower than shared memory within one server). In HPC, high speed interconnect and specialized RDMA-aware communication libraries make distributed parallel computation very scalable. Grex uses InfiniBand interconnect (IB).
Most often (but not always), parallel programs are built upon a low-level message passing library called MPI. Refer to the Software section for more information about parallel libraries on Grex. Examples of distributed parallel codes are GAMESS-US, ORCA , LAMMPS , VASP , … etc.
The distributed parallel jobs can be placed across their compute nodes in several ways (i.e., how many parallel tasks per compute node?). Thus, SLURM resource request syntax allows to specify the required layout of nodes/tasks (or nodes/tasks/threads, or even nodes/tasks/GPUs since hybrid MPI+OpenMP and MPI+GPU programs exist). A consideration about the layout is a tradeoff between making the program work faster (and sometimes to work correctly at all) and making the scheduler’s work easier.
A well written MPI software theoretically should not care how the tasks are distributed across how many physical compute nodes. Thus, SLURM’s --ntasks= request (similar to the old Torque procs=) specified without --nodes would work and make the scheduling easier.
A note on process starting:
Since MPI jobs are distributed, there should be a mechanism to start the compute processes across all of the nodes (or CPUs) allocated for it. The mechanism should know which nodes to use, and how many. Most modern MPI implementations “tightly integrate” with SLURM, so they will get this information automatically via a Process Management Interface (PMI). SLURM provides its own job starting command called srun . Most MPI implementations also provide their own job spawned commands, usually called mpiexec or mpirun. These are specific to each MPI vendor/kind and not well standardized, and differ in the support of SLURM.
For example, OpenMPI (the default, supported MPI implementation) on Grex is compiled against PMIx (3.x, 4.x) or PMI1 (1.6.5). So, it is preferable to use srun instead of mpiexec to kick start the MPI processes, because srun would use PMI.
For Intel MPI (another MPI, also available on Grex and required by some of the binary codes, like ANSYS or ADF), srun sometimes may not work, but PMI1 can be used with mpiexec.hydra by setting the following environment variable:
export I_PMI_LIBRARY=/opt/slurm/lib/libpmi.soSome examples#
Here is an example for running MPI job (in this case, Quantum ESPRESSO) using 32 cores:
#!/bin/bash
#SBATCH --time=0-8:00:00
#SBATCH --mem-per-cpu=1500M
#SBATCH --ntasks=32
#SBATCH --job-name="QE-Job"
# A example of an MPI parallel that
# takes 32 cores on Grex for 8 hours.
# Load the modules:
module load arch/avx512 intel/2023.2 openmpi/4.1.6 espresso/7.3.1
export OMP_NUM_THREADS=1
echo "Starting run at: `date`"
srun pw.x -in MyFile.scf.in > Myfile.scf.log
echo "Job finished with exit code $? at: `date`"
However, in practice there are cases when layout should be more restrictive. If the software code assumes equal distribution of processes per node, the request should be --nodes=N --ntasks-per-node=M. A similar case is MPMD codes (Like NWCHem or GAMESS-US or OpenMolcas) that have some of the processes doing computation and some communication functions, and therefore requires at least two tasks running per each node.
For some codes, especially for large parallel jobs with intensive communication between tasks there can be performance differences due to memory and interconnect bandwidths, depending on whether the same number of parallel tasks is compacted on few nodes or spread across many of them. Find an example of the job below.
#!/bin/bash
#SBATCH --nodes=4
#SBATCH --ntasks-per-node=8
#SBATCH --mem-per-cpu=4000M
#SBATCH --job-name="NWchem-Job"
#SBATCH --job-name=NWchem-dft-test
# Adjust the number of tasks, time and memory required.
# the above spec is for 32 compute tasks over 4 nodes.
# Load the modules:
module load intel/15.0.5.223 ompi/3.1.4 nwchem/6.8.1
echo "Starting run at: `date`"
which nwchem
# Uncomment/Change these in case you want to use custom basis sets
NWCHEMROOT=/global/software/cent7/nwchem/6.8.1-intel15-ompi314
export NWCHEM_NWPW_LIBRARY=${NWCHEMROOT}/data/libraryps
export NWCHEM_BASIS_LIBRARY=${NWCHEMROOT}/data/libraries
# In most cases SCRATCH_DIR would be on local nodes scratch
# While results are in the same directory
export NWCHEM_SCRATCH_DIR=$TMPDIR
export NWCHEM_PERMANENT_DIR=`pwd`
# Optional memory setting; note that this one or the one in your code
# must match the #SBATCH --mem-per-cpu times compute tasks !
export NWCHEM_MEMORY_TOTAL=2000000000 # 24000 MB, double precision words only
export MKL_NUM_THREADS=1
srun nwchem dft_feco5.nw > dft_feco5.$SLURM_JOBID.log
echo "Program finished with exit code $? at: `date`"
OpenMPI#
OpenMPI is the default MPI implementation for Grex (and Compute Canada, now the Alliance). The modules for it on Grex are called ompi . The MPI example scripts above are all OpenMPI based. The old version 1.6.5 is there for compatibility reasons with older software; most users should use 3.1.x or 4.x.x versions. Using srun is recommended in all cases.
Intel MPI#
For applications using IntelMPI (impi modules on Grex, or Intel-MPI based software from Compute Canada CVMFS software stack), a few environment variables have to be set. The following link explains it: Using SLURM with PMI .
The JLab documentation example shows an example of SLURM script with IntelMPI .
Other MPIs#
Finally, some canned codes like ANSYS or StatCCM+ would use a vendor-specific MPI implementation that would not tightly integrate with our scheduler’s process to CPU core placement. In that case, several whole nodes (that is, with the number of tasks equal to the node’s number of CPU cores) should be requested to prevent the impact on other jobs with resource congestion.
Such codes will also require a nodelist (machinefile) file obtained from SLURM and provided to them in their own format.
A custom script slurm_hl2hl.py makes this easier (see CC StarCCM+ or CC ANSYS documentation ). The script slurm_hl2hl.py is already available on Grex.
slurm_hl2hl.py --format STAR-CCM+ > machinefileJob arrays#
Job arrays allow for submitting many similar jobs “in one blow”. It saves users work on job submission, and also makes SLURM scheduler more efficient in scheduling the array jobs because it would know they are the same with respect to size, expected wall time etc.
Array jobs work most naturally when a single code has to be applied for parameter sweep and/or to a large number of input files that are regularly named, for example as: test1.in, test2.in, … test99.in
Then, a single job script with #SBATCH --array=1,99 can be used to submit the 99 jobs.
In order to distinguish between the input files, within each of the jobs at run time, you would have to obtain a value for the array index. Which is set by SLURM as ${SLURM_ARRAY_TASK_ID} environment variable. The call to the code on a particular input will then be like:
./my_code test${SLURM_ARRAY_TASK_ID}.inThis way each of the array element jobs can distinguish their own portion of the work to do. A real life example is below; it attempts to run all of the Gaussian standard tests which have names of the format test0001.com, test0002.com, .. test1204.com, etc. Note the printf trick to deal with trailing zeroes in the input names.
#!/bin/bash
#SBATCH --cpus-per-task=1
#SBATCH --mem=1000MB
#SBATCH --job-name="G16-tests"
#SBATCH --array=1-1204
echo "Current working directory is `pwd`"
echo "Running on `hostname`"
echo "Starting run at: `date`"
# Set up the Gaussian environment using the module command:
module load gaussian/g16.c01
# Run g16 on an array job element
id=`printf "%04d" $SLURM_ARRAY_TASK_ID`
v=test${id}.com
w=`basename $v .com`
g16 < $v > ${w}.${SLURM_JOBID}.out
echo "Job finished with exit code $? at: `date`"
There are limits on how large array jobs can be (see our scheduling policies): the maximal number of elements in job array, as well as the maximal number of jobs that can be submitted by a user.
Using CC CVMFS software#
As explained in more detail in the software/Modules documentation, we provide Compute Canada’s software environment. Most of it can run out of the box by just specifying the corresponding module.
There are some caveats:
Some of the Compute Canada software might have hardcoded environment variables that exist only on these systems. An example is SLURM_TMPDIR. On Grex, add export SLURM_TMPDIR=$TMPDIR to your job scripts.
In general, it is hard to containerize HPC. So the software that requires low-level hardware/device drivers access (OpenMPI, CUDA) may have problems when running on non-CC systems. Newer version of OpenMPI (3.1.x) seems to be more portable for using the PMIx job starting mechanism.
“Restricted” (commercial) software’s binaries are not distributed by Compute Canada CVMFS due to the obvious licensing issues. It has to be installed locally on Grex.
Having said that, module load CCEnv gives the right software environment to be run on Grex for a vast majority of threaded and serial software items from CC software stack. See discussion about MPI-parallel jobs below.
Because of the distributed nature of CVMFS, it might take time to download a program or library or data file. It would probably make sense to first access it interactively or from an interactive job to warm the CVMFS local cache, to avoid job failures due to the delay.
Below is an example of an R serial job that uses quite a few packages from Compute Canada software stack.
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH --mem-per-cpu=4000M
#SBATCH --time=0-72:00:00
#SBATCH --job-name="R-gdal-jags-bench"
cd ${SLURM_SUBMIT_DIR}
# Load the modules:
module load CCEnv
module load arch/avx512
module load StdEnv/2023
module load gcc/12.3 r/4.4.0 jags/4.3.2 geos/3.12.0 gdal/3.9.1
export MKL_NUM_THREADS=1
echo "Starting run at: `date`"
R --vanilla < Benchmark.R &> benchmark.${SLURM_JOBID}.txt
echo "Program finished with exit code $? at: `date`"
Users of contributed systems which are newer than the original Grex nodes might want to switch to arch/avx2 or arch/avx512 from the default arch/sse3.
Using CC CVMFS software that is MPI-based.#
We have found that the recent Compute Canada toolchains that use OpenMPI 3.1.x work on Grex without any changes (that is, with srun). Therefore, for OpenMPI based applications, we recommend to load Compute Canada’s software that depends on the recent toolchains, 2018.3 or later (Intel 2018 compilers, GCC 7.3 compilers and openmpi/3.1.2).
For example, the module commands below would load the Intel/OpenMPI 3.1.2 toolchain-based environment:
module load CCEnv
module load StdEnv/2018.3Below is an arbitrarily chosen IMB benchmark result for MPI1 on Grex, the sendrecv tests using two processes on two nodes with several MPI implementations (CC means MPI coming from the Compute Canada (now, the Alliance) stack, Grex means compiled locally on Grex).
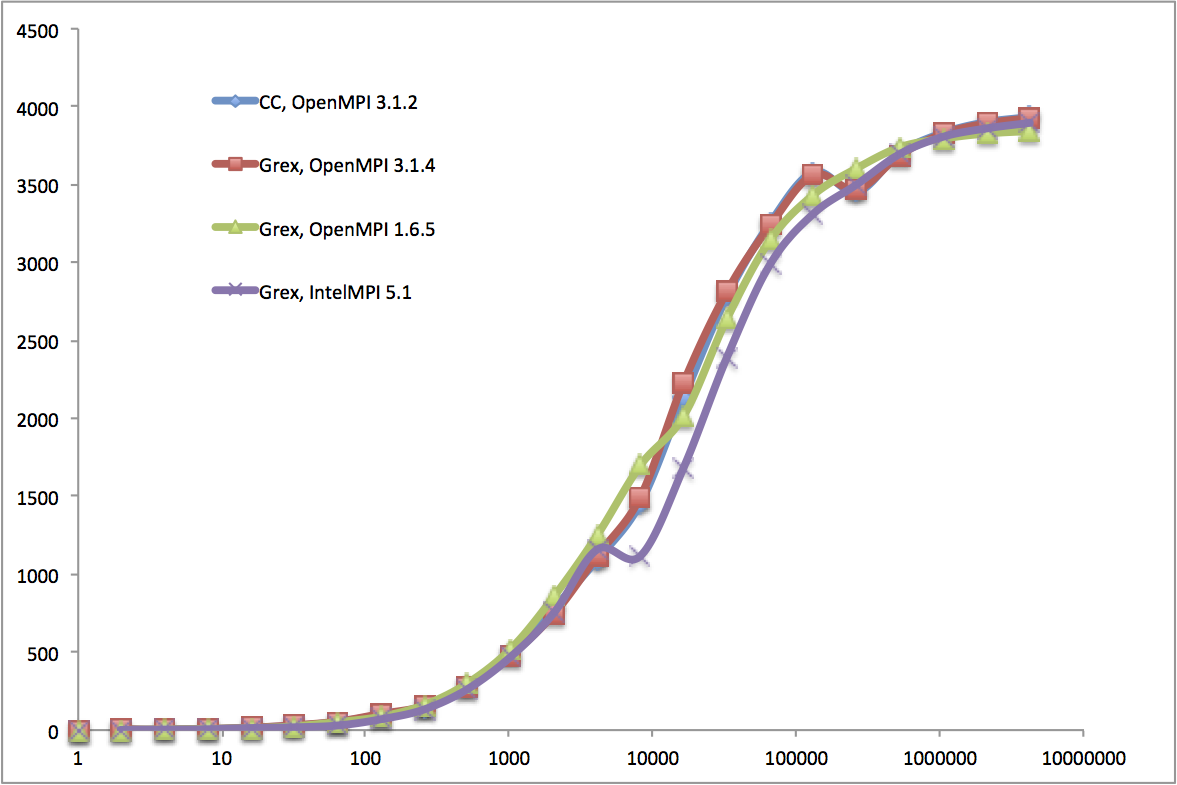
You can see that differences in performance between OpenMPI 3.1.x from CC stack and Grex are minor for this benchmark, even without attempting any local tuning for the CC OpenMPI.
Using New 2020 CC CVMFS Stack#
Since Spring 2021, Compute Canada has updated the default software stack on their CVMFS distribution to StdEnv/2020 and gentoo. This version will not run on legacy Grex partitions (compute) at all, because it requires AVX2 CPU architecture. It will work as expected on all new GPU and CPU nodes (skylake, largemem, gpu and contributed systems).